 English
English 简体中文
简体中文



PCR实验污染的强势解决方案!实用贴!!!
时间:2018-05-02 来源:EASTWIN浏览次数:189
(一) 查找污染源
如果不慎发生污染情况,应从下面几条出发,逐一分析,排除污染。
1、设立阴阳性对照:有利于监测反应体系各成分的污染情况。选择阳性对照时,应选择扩增弱,且重复性好的样品,因强阳性对照可产生大量不必要的扩增序列,反而可能成为潜在的污染源。如果以含靶序列的重组质粒为对照,100 个拷贝之内的靶序列就足以产生阳性扩增。阴性对照的选择亦要慎重,因为PCR 敏感性极高,可以从其它方法(Sourthern 印迹或点杂交等)检测阴性的标本中检测出极微量的靶分子。此外,每次扩增均应包括PCR 体系中各试剂的时机对照,即包括PCR 反应所需的全部成分,而不加模板DNA,这对监测试剂中PCR 产物残留污染是非常有益的。如果扩增结果中试剂对照为阳性结果,就是某一种或数种试剂被污染了。此时,要全部更换一批新的试剂进行扩增,扩增时设立不同的反应管,每一管含有一种被检测试剂,在检出污染试剂后,应马上处理。
(二) 环境污染
在排除试剂污染的可能性外,更换试剂后,若不久又发现试剂被污染了,如果预防措施比较严密,则考虑可能为环境污染。环境污染中常见的污染源主要有:
1. 模板提取时真空抽干装置;
2. 凝胶电泳加样器;
3. 电泳装置;
4. 紫外分析仪;
5. 切胶用刀或手术刀片;
6. 离心机;
7. 冰箱门把手,冷冻架,门把手或实验台面等;
此时可用擦拭实验来查找可疑污染源:
1)用无菌水浸泡过的灭菌棉签擦拭可疑污染源;
2)0.1ml 去离子水浸泡;
3)取5ml 做PCR 实验;
4)电泳检测结果。
8. 气溶胶。如果经过上述追踪实验,仍不能查找到确切污染源,则污染可能是由空气中PCR 产物的气溶胶造成的,此时就应该更换实验场所,若条件不允许,则重新设计新的引物(与原引物无相关性)。
(三) 污染处理
环境污染
1. 稀酸处理法:对可疑器具用1mol/L 盐酸擦拭或浸泡,使残余DNA 脱嘌呤; 2. 紫外照射(UV)法:紫外波长(nm)一般选择254/300nm,照射30min 即可。需要注意的是,选择UV 作为消除残留PCR 产物污染时,要考虑PCR 产物的长度与产物序列中碱基的分布,UV 照射仅对500bp 以上长片段有效,对短片段效果不大。UV 照射时,PCR产物中嘧啶碱基会形成二聚体,这些二聚体可使延伸终止,但并不是DNA 链中所有嘧啶均能形成二聚体,且UV 照射还可使二聚体断裂。形成二聚体的程度取决于UV 波长,嘧啶二聚体的类型及与二聚体位点相邻核苷酸的序列。在受照射的长DNA 链上,形成二聚体缺陷的数量少于0.065/碱基,其他非二聚体的光照损伤(如环丁烷型嘧啶复合体,胸腺嘧啶乙二醇,DNA 链间与链内的交联和DNA 断裂等)均可终止Taq DNA 聚合酶的延伸。这些位点的数量与二聚体位点相当。如果这些位点( 0.13/碱基)在DNA 分子上随机分布,一个500bp片段的DNA 分子链上将有32 处损伤位点,那么,105个这样的分子中每个分子中会至少有一处损伤。相反,如果100bp 的片段,每条链上仅有6 处损伤,105个拷贝分子中将有许多分子没有任何损伤。这就是UV 照射有一定的片段长度限制的原因
可采用下列方法之一处理:
1. DNase I 法:PCR 混合液(未加模板和Taq 聚合酶)加入0.5U DNase I,室温反应30min 后加热灭活,然后加入模板和Taq 聚合酶进行正常PCR 扩增。该方法的优点是不需要知道污染DNA 的序列;
2. 内切酶法:选择识别4 个碱基的内切酶(如Msp I 和Taq I 等),可同时选择几种,以克服用一种酶只能识别特定序列的缺陷,室温作用1h 后加热灭活进行PCR; 3. 紫外照射法:未加模板和Taq 聚合酶的PCR 混合液进行紫外照射,注意事项与方法同上述UV 照射法;
4. g 射线辐射法:1.5kGy 的辐射可完全破坏0.1ng 基因组DNA,2.0 kGy 可破坏104 拷贝的质粒分子,4.0 kGy 仍不影响PCR,但高于此限度会使PCR 扩增效率下降。引物可受照射而不影响PCR,g 射线是通过水的离子化产生自由基来破坏DNA 的。
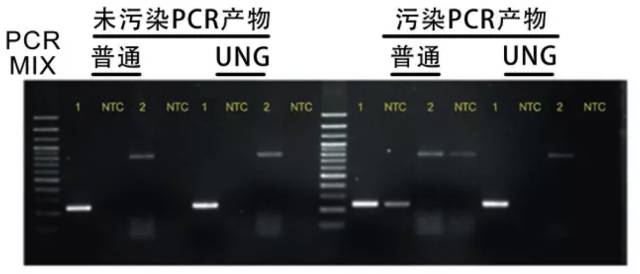
由于UV 照射的去污染作用对500bp 以下的片段效果不好,而临床用于检测的PCR 扩增片段通常为300bp 左右,因此UNG 的预防作用日益受到重视和肯定。
1. 原理:在PCR 产物或引物中用dU 代替dT。这种dU 化的PCR 产物与UNG 一起孵育,因UDG 可裂解尿嘧啶碱基和糖磷酸骨架间的N-糖基键,可除去dU 而阻止TaqDNA 聚合酶的延伸,从而失去被再扩增的能力。UNG 对不含dU 的模板无任何影响。UNG 可从单或双链DNA 中消除尿嘧啶,而对RNA 中的尿嘧啶和单一尿嘧啶分子则无任何作用。
2. dUTP 法:用dUTP 代替dTTP,使产物中掺入大量dU。在再次进行PCR 扩增前,用UNG 处理PCR 混合液即可消除PCR 产物的残留污染。由于UNG 在PCR 循环中的变性一步便可被灭活,因此不会影响含dU 的新的PCR 产物。
3. dU 引物法:合成引物时以dU 代dT,这样PCR 产物中仅5ˊ端带dU。UNG 处理后,引物失去了结合位点而不能扩增。对长片段(1-2kb 以上)的扩增用dUTP 法效率较用dTTP低,而用dU 法就可克服这一缺点。dU 引物最好将dU 设计在3ˊ端或近ˊ端。该法仅能用于引物以外试剂的处理。
4. 优点:可以去除任何来源的污染;UNG 处理可以和PCR 扩增在同一个反应管内进行;由于扩增产物中有大量dU 存在,可彻底消除污染源。
5. 需注意的是掺入dUTP 的DNA不应对产物的任何操作有影响,在进行PCR 产物克隆时,应该转化UNG-(UNG 缺陷)大肠杆菌受体菌,否则转化产物会被受体菌UNG 消
化掉。
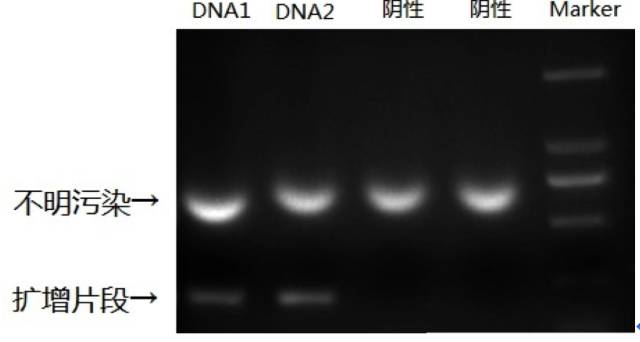
用于去除标本中污染的核酸和杂质,原理如下:
1)用一生物素标记的单链RNA 探针与待扩核酸杂交,杂交区域是非扩增区;
2)用包被链霉亲和素的固相载体来捕获带有生物素探针的杂交核酸,通过漂洗可去除污染的扩增产物和杂质;
3)洗脱靶分子后用特异引物扩增非RNA 探针杂交区域。第2)步的漂洗后可用PCR 检测以确定标本是否被扩增产物或重组质粒污染。
也称为链特异性PCR,主要指用于RNA 模板的特异性PCR 法,该法可明显降低假阳性而不影响PCR 的敏感性。其关键在于设计引物,逆转录引物的3ˊ端(A 区)有2 0 个核苷酸左右为模板的特异性互不序列,5ˊ端2 0 个核苷酸(C 区)为附加修饰碱基。与mRNA逆转录后,经超速离心使cDNA 与多余引物分开,再用和第二引物(C)以第一链cDNA为模板合成第二链cDNA,以后的PCR 循环中用逆转录引物的B 区和引物C 进行扩增加尾cDNA,而污染的DNA 或质粒DNA 才不会被扩增。
该对引物扩增时通过病毒DNA克隆如入质粒的位点。这一区域只存在完整的原病毒中,在重组质粒中,这一区域分成两个区域与克隆位点被。如果重组质粒污染了标本,也不能扩增出任何条带,即使出现了扩增带,其大小也与预期的不同。只有原病毒DNA 才能被引物扩增,因此只要出现预期大小的扩增带就可以证明标本是阳性的,该法试用于环状靶分子系列。
(声明:内容来自网络,仅供分享之用,版权属于原作者。如涉及版权问题,请与小编联系。)


